
Transannular C–H functionalization of cycloalkane carboxylic acids
Inventors & their inventions
Axial: https://linktr.ee/axialxyz
Axial partners with great founders and inventors. We invest in early-stage life sciences companies such as Appia Bio, Seranova Bio, Delix Therapeutics, Simcha Therapeutics, among others often when they are no more than an idea. We are fanatical about helping the rare inventor who is compelled to build their own enduring business. If you or someone you know has a great idea or company in life sciences, Axial would be excited to get to know you and possibly invest in your vision and company. We are excited to be in business with you — email us at info@axialvc.com
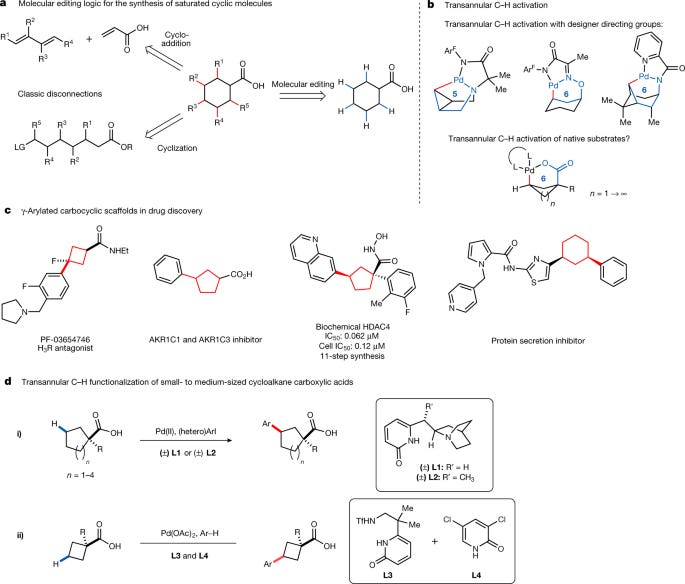
This paper details the development of novel catalytic methods for the site- and diastereoselective synthesis of functionalized carbocycles through transannular C-H functionalization of cycloalkane carboxylic acids. Carbocycles, cyclic structures containing only carbon atoms, are prevalent structural motifs in both natural products and pharmaceuticals. Their inherent rigidity and control over molecular shape contribute significantly to the biological activity and bioavailability of drug candidates. Traditional synthetic approaches to carbocycles often involve multi-step processes, including cycloadditions or intramolecular cyclizations from acyclic precursors, which can be challenging and inefficient. This study proposes C-H activation as a more direct and potentially efficient strategy for the synthesis of these important compounds.
The central challenge in achieving transannular C-H functionalization lies in the inherent strain associated with forming the required palladacycles, especially with larger rings. Existing methods often rely on pre-installed directing groups or are limited to specific ring sizes and functional groups. This research overcomes these limitations by introducing two novel classes of ligands: quinuclidine-pyridones (QuinNuPyridones) and sulfonamide-pyridones (SulfonaPyridones). These ligands facilitate the selective transannular γ-C-H arylation of a wide range of cycloalkane carboxylic acids, encompassing ring sizes from cyclobutane to cyclooctane.
The researchers demonstrate the effectiveness of QuinNuPyridones, specifically L1 and L2, in promoting the transannular γ-arylation of cyclopentane to cyclooctane carboxylic acids. Remarkably, excellent γ-regioselectivity is observed even in the presence of multiple competing β-C-H bonds. This high level of regiocontrol is a significant advancement in C-H activation methodologies. The reaction conditions were optimized using a combination of palladium catalyst, ligand, oxidant, and solvent. The scope of the reaction was explored by varying both the substituents on the cycloalkane ring and the aryl iodide coupling partner. A diverse range of substituents, including electron-donating and electron-withdrawing groups, are well tolerated, showcasing the broad applicability of the method. The utility of this methodology is further highlighted by the successful synthesis of several patented biologically active molecules in just two steps. Previous syntheses of these compounds often required considerably more steps (up to eleven in one case).
The successful transannular γ-arylation of cyclobutane carboxylic acids proved to be more challenging due to the greater strain and preference for β-functionalization. However, the researchers addressed this challenge by employing a different ligand system: the SulfonaPyridone ligand L3 in combination with a monodentate pyridone ligand L4. This combination enabled the highly selective transannular γ-arylation of cyclobutane carboxylic acids via a double C-H activation pathway. This strategy highlights the critical role of ligand design in controlling both reactivity and selectivity in C-H activation reactions. The precise mechanism by which L3 and L4 achieve this high level of selectivity is not fully elucidated, but the authors propose that the combination of these ligands may promote a sequential process involving non-directed C-H activation of the arene and then a subsequent C-H activation/cross-coupling on the cyclobutane ring.
The authors thoroughly investigate the reaction scope for both the cyclopentane-cyclooctane and cyclobutane systems. For the larger rings (cyclopentane to cyclooctane), a wide range of substrates with various α-substituents were successfully arylated, demonstrating the tolerance of the reaction for steric hindrance. Both electron-rich and electron-poor aryl iodides and even heteroaryl iodides reacted efficiently. For cyclobutane carboxylic acids, a similar scope of arene coupling partners is demonstrated although the yield varied depending on the electronics of the arene. The authors further demonstrated that the methodology could be applied to complex molecules, including the late-stage functionalization of the natural product isosteviol.
Beyond the synthesis of novel compounds, the research contributes significantly to the understanding of C-H activation catalysis. The observed regioselectivity and the impact of ligand structure on the outcome of the reaction provide valuable insights into the mechanistic details of transannular C-H activation. The observation that different ligand systems are required for optimal results with different ring sizes further underscores the importance of ligand design in overcoming the challenges of strain and selectivity in these transformations.
In summary, this work represents a significant advancement in the field of C-H activation catalysis. The development of new ligand systems that enable the highly selective transannular γ-arylation of cycloalkane carboxylic acids, particularly the challenging cyclobutane system, opens up new possibilities for the synthesis of complex carbocycles. The methodology is remarkably versatile, tolerating a wide range of substrates and coupling partners, and offers a simplified and efficient route to biologically relevant molecules. The efficient synthesis of known biologically active compounds in fewer steps than previous approaches showcases the practical value and broad applicability of this novel catalytic system. This research not only provides new tools for organic synthesis but also deepens our understanding of the factors that govern the selectivity and reactivity of C-H activation reactions. The insights gained could inspire the design of future catalyst systems for a wider range of C-H functionalization transformations. Future research could focus on expanding the scope of the reaction to other functional group transformations, exploring the catalytic mechanism in more detail, and applying the methodology to the synthesis of even more complex and biologically important molecules.
https://www.nature.com/articles/s41586-023-06000-z
