

Perspective on the Current State-of-the-Art of Quantum Computing for Drug Discovery Applications
Perspective on the Current State-of-the-Art of Quantum Computing for Drug Discovery Applications
Inventors & their inventions
Axial: https://linktr.ee/axialxyz
Axial partners with great founders and inventors. We invest in early-stage life sciences companies such as Appia Bio, Seranova Bio, Delix Therapeutics, Simcha Therapeutics, among others often when they are no more than an idea. We are fanatical about helping the rare inventor who is compelled to build their own enduring business. If you or someone you know has a great idea or company in life sciences, Axial would be excited to get to know you and possibly invest in your vision and company. We are excited to be in business with you — email us at info@axialvc.com
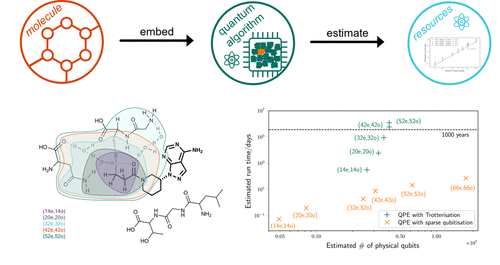
The paper presents a perspective on the potential for quantum computing to enable more accurate computational chemistry calculations that are relevant for pharmaceutical applications. It focuses specifically on the ability of quantum computers to perform complete active space configuration interaction (CASCI) calculations to find the ground state energy of molecular systems, which can currently only be done for relatively small active spaces on classical computers due to the exponential scaling.
The introduction provides useful background on quantum chemistry methods and the importance of being able to perform accurate calculations on strongly correlated systems which are difficult to treat with standard approximate methods like density functional theory. It makes the case that while formal definitions of "quantum advantage" are difficult to apply usefully to chemistry, a working definition of "quantum benefit" as being able to outperform classical computational methods for industrially relevant chemical problems is more practical.
The paper then reviews the basics of mapping the electronic structure problem onto a quantum computer, representing the molecular Hamiltonian in terms of qubits using techniques like the Jordan-Wigner or Bravyi-Kitaev transformations. It outlines the two main quantum algorithms for calculating molecular energies - the variational quantum eigensolver (VQE) and quantum phase estimation (QPE). The authors argue that QPE is likely to be favored over VQE for large active spaces due to its more favorable scaling, which motivates their focus on QPE for the remainder of the paper.
A key component is estimating the resources required to run QPE for realistic chemical systems. This depends on finding an efficient way to implement the time-evolution operator U = e-iHt on the quantum computer. The authors compare two methods - Trotterization and qubitization. Trotterization uses a approximation of U by breaking it into many short time steps, requiring very deep circuits that scale exponentially with the simulation time. The qubitization approach instead encodes the Hamiltonian into an auxiliary state and allows implementing U much more efficiently, dramatically reducing the circuit depth and coherence time requirements.
To illustrate the potential, the authors consider the pharmaceutical drug Ibrutinib bound to the Bruton's tyrosine kinase protein. They estimate the resources needed to fully quantum simulate progressively larger embedding regions around the binding pocket using active spaces up to around 50 orbitals and electrons. Their key finding is that with the qubitization approach and recent algorithmic improvements, this type of simulation could potentially be carried out on a large error-corrected quantum computer in just a few days of runtime. In contrast, their estimates using Trotterization show runtimes of over 1000 years would be required.
While many challenges remain in building a large fault-tolerant quantum computer capable of these calculations, the paper paints an exciting picture of the potential for quantum computing to enable currently intractable quantum chemistry calculations that could be transformative for pharmaceutical research. It highlights how the field is progressing rapidly in terms of hardware, error correction, and algorithms for quantum simulation.
https://pubs.acs.org/doi/10.1021/acs.jctc.2c00574
