


In vivo photoreceptor base editing ameliorates rhodopsin-E150K autosomal-recessive retinitis pigmentosa in mice
Inventors & their inventions
Axial: https://linktr.ee/axialxyz
Axial partners with great founders and inventors. We invest in early-stage life sciences companies such as Appia Bio, Seranova Bio, Delix Therapeutics, Simcha Therapeutics, among others often when they are no more than an idea. We are fanatical about helping the rare inventor who is compelled to build their own enduring business. If you or someone you know has a great idea or company in life sciences, Axial would be excited to get to know you and possibly invest in your vision and company. We are excited to be in business with you — email us at info@axialvc.com
This study investigates a potential gene therapy treatment for retinitis pigmentosa (RP) caused by a specific mutation in the rhodopsin gene. The researchers focused on correcting the E150K mutation, which causes autosomal recessive RP, using CRISPR-based precision editing techniques in mice.
Rhodopsin plays a dual role in rod photoreceptors - it acts both as a light sensor and as a crucial structural protein. In rod cells, rhodopsin makes up over 90% of the protein content in the outer segment disc membranes and occupies about 50% of the surface area. Each rod cell contains approximately 80,000 rhodopsin molecules per disc, totaling around 400 trillion molecules per eye. The protein undergoes rapid turnover, with the entire rod outer segment being replaced approximately every 10 days in rodents and primates. This makes sustained high expression of rhodopsin essential for maintaining both the structure and function of rod photoreceptors.
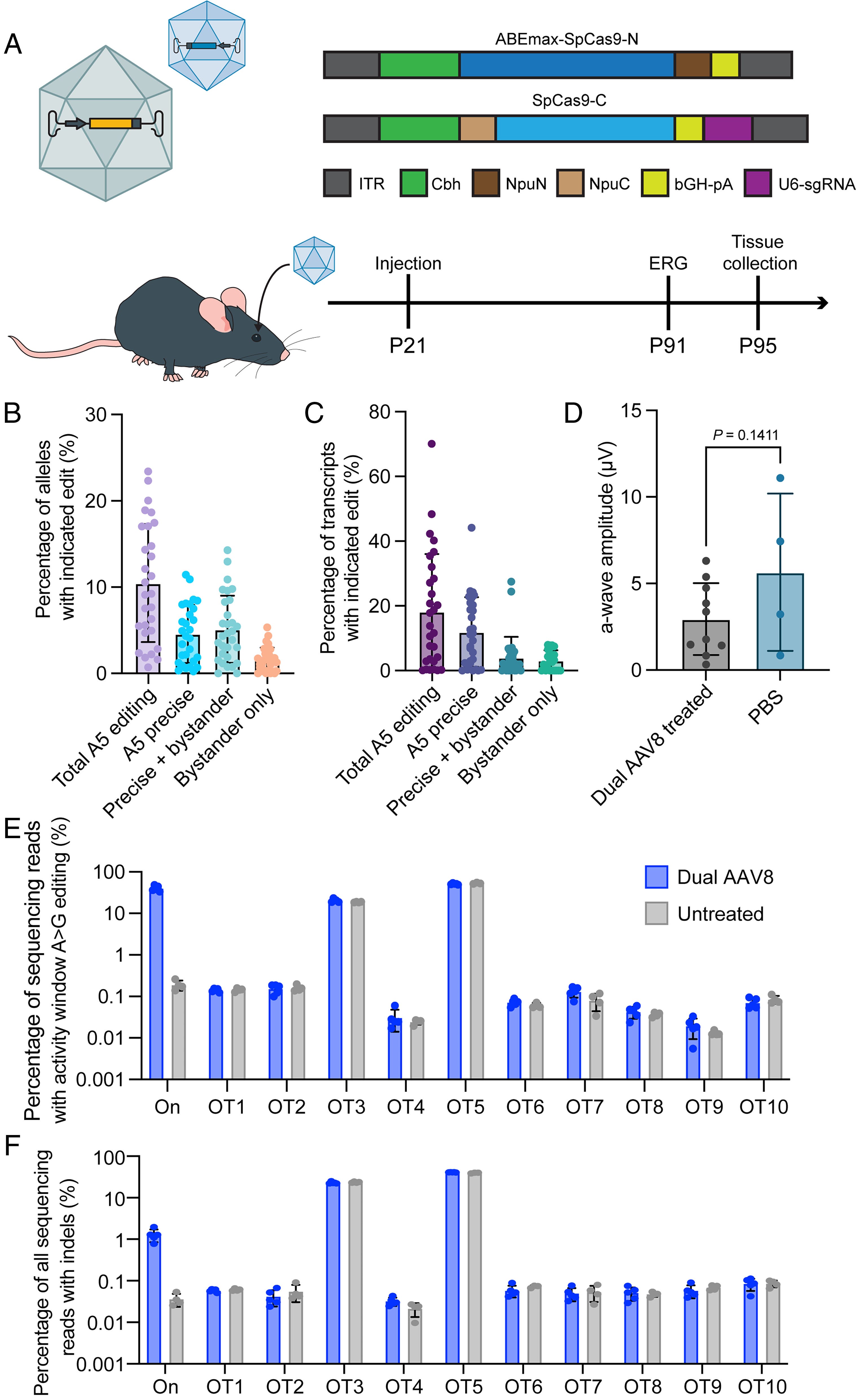
The researchers used CRISPR/Cas9-derived adenine base editing (ABE) to correct the G>A mutation that causes the E150K variant. They first developed and tested their editing strategy in a modified HEK293T cell line carrying the rhodopsin mutation. After screening various guide RNAs and base editor variants, they found that ABEmax combined with a specific guide RNA (A5) provided the best balance of editing efficiency and precision, achieving about 12% total editing and 9% precise correction of the target base.
One challenge with base editing was the occurrence of "bystander editing" - unintended editing of other adenine bases near the target site. To determine whether these bystander edits would affect protein function, the researchers expressed and characterized all possible rhodopsin variants that could result from the editing process. They tested ten different variants, including wild-type rhodopsin, the E150K mutant, and eight potential bystander-edited versions. Importantly, all variants showed normal spectral properties when reconstituted with the visual chromophore 11-cis-retinal, and all maintained the ability to activate transducin (the G-protein involved in phototransduction) at levels similar to wild-type rhodopsin.
The researchers also developed an alternative prime editing strategy that could potentially offer greater precision without bystander editing. While they achieved some success with this approach in vitro, they ultimately proceeded with the base editing strategy for in vivo studies due to its higher efficiency.
For delivery of the base editor to mouse retinas, the team developed a dual AAV system where the base editor was split between two viral vectors and could be reconstituted in cells that received both components. They first treated mice at postnatal day 21 (P21) with subretinal injections of the dual AAV system. Analysis after 10 weeks showed encouraging editing rates, with up to 44% correction of the mutation in RNA transcripts. However, they did not observe significant rescue of retinal function as measured by electroretinography (ERG).
The researchers hypothesized that the timing of treatment might be crucial, as the Rho-E150K mice lose over 20% of their photoreceptors by postnatal day 30 and about 60% by two months of age. They therefore tried treating mice earlier, at postnatal day 15 (P15). This earlier intervention proved more successful, with treated animals showing preserved ERG responses at 14 weeks post-treatment while untreated animals lost nearly all photoreceptor function. Histological analysis confirmed that treated retinas maintained more photoreceptor nuclei in the outer nuclear layer compared to untreated controls.
Immunohistochemistry performed 15 weeks after treatment showed that the therapy restored rhodopsin expression in the outer segments of treated retinas, while untreated mutant retinas showed virtually no rhodopsin expression. The rescue effect was not uniform across the retina, with some areas showing strong preservation of photoreceptors and others showing little to no effect, likely due to incomplete viral delivery or expression patterns.
The study highlights both the promise and challenges of gene editing approaches for treating inherited retinal diseases, particularly those affecting proteins with both structural and functional roles. The researchers note that the timing of intervention appears crucial for therapeutic success, as early treatment (P15) showed benefits while later treatment (P21) did not, despite achieving similar editing efficiencies.
The massive abundance of rhodopsin in rod cells presents a particular challenge for gene therapy. Each rod contains over 64 million rhodopsin molecules, with about 10% being turned over daily. This requires the continuous production of thousands of new rhodopsin molecules per cell per day. The researchers calculate that each rod generates 3,400-5,000 rhodopsin mRNA molecules daily, or about one new transcript every 25 seconds. In a mouse retina with 6.4 million rods, this means approximately 250,000 new mRNA molecules must be generated every second to maintain normal rhodopsin levels.
The study also raises important considerations for clinical translation. The E150K mutation is relatively rare, which could make it challenging to conduct clinical trials and obtain regulatory approval. The researchers suggest that innovative approaches combining complementary therapies to extend the therapeutic window, or modified regulatory frameworks for rare mutations, might be necessary to bring such treatments to patients.
The mechanisms of retinal degeneration in E150K-associated RP remain incompletely understood. While the mutation may affect protein trafficking and oligomerization, the researchers found that bystander-edited variants remained functional in biochemical assays. They hypothesize that removing the positive charge at position 150 (which occurs in most editing outcomes) might be sufficient to prevent degeneration, as the lysine at this position may disrupt normal protein packing in the rod outer segment.
This work demonstrates that precision gene editing can partially rescue retinal structure and function in a mouse model of RP, but the degree of rescue appears highly dependent on treatment timing and may require optimization of delivery methods to achieve more uniform correction across the retina. The study provides important insights for the development of gene editing therapies for inherited retinal diseases, particularly those affecting proteins with both structural and signaling functions.
https://www.pnas.org/doi/full/10.1073/pnas.2416827121
