


Axial: https://linktr.ee/axialxyz
Axial partners with great founders and inventors. We invest in early-stage life sciences companies such as Appia Bio, Seranova Bio, Delix Therapeutics, Simcha Therapeutics, among others often when they are no more than an idea. We are fanatical about helping the rare inventor who is compelled to build their own enduring business. If you or someone you know has a great idea or company in life sciences, Axial would be excited to get to know you and possibly invest in your vision and company. We are excited to be in business with you — email us at info@axialvc.com
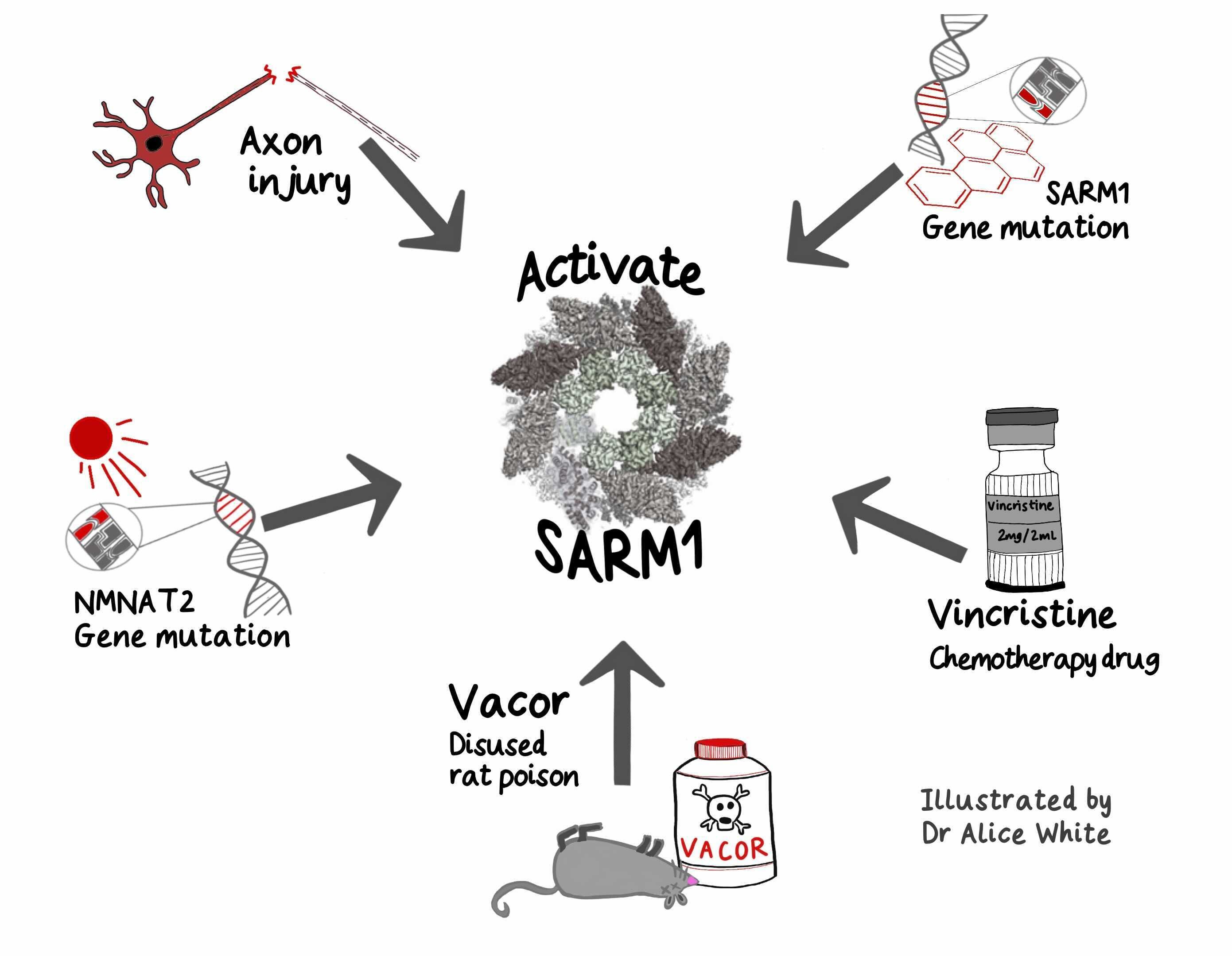
SARM1 has emerged as a promising therapeutic target for preventing axonal degeneration associated with peripheral neuropathy, traumatic brain injury (TBI), and potentially even neurodegenerative diseases like Parkinson's disease. As well as a central executor of the Wallerian degeneration pathway. Inhibiting SARM1 activity could completely change the disease trajectory for millions of patients suffering from these conditions.
Peripheral neuropathies resulting from physical injury, metabolic disorders like diabetes, infectious diseases, autoimmune disorders, nutrient deficiencies, or side effects of chemotherapy drugs are estimated to impact over 20M people in the US alone. By blocking the downstream consequences of axon damage via SARM1 inhibition, peripheral neuropathy progression could be halted irrespective of the root cause.
As an enzyme possessing intrinsic NAD+ hydrolase activity, the active site of SARM1 is druggable by small molecules that occupy the NAD+ binding pocket and block access to the substrate. Lead candidates would need to demonstrate high potency and selectivity for SARM1 over other NAD+ consuming enzymes, favorable pharmacokinetic properties enabling sufficient nerve tissue exposure, and a clean safety profile devoid of mechanism-based toxicities. Local delivery methods directly to affected nerves could also help minimize any on-target effects in immune regulation or other physiological roles of SARM1.
Gene therapy offers another potential approach for sustainable SARM1 inhibition within peripheral nerves. In fact, localized injection of adeno-associated viruses encoding dominant negative SARM1 suppressed Wallerian degeneration for over 10 days in mice - comparable efficacy to genetic SARM1 deletion. This suggests durable axon protection is achievable via gene therapy, but safety and manufacturability of gene therapies remains a key consideration.
The acute axonal damage and degeneration precipitated by traumatic brain injury drives much of its long term morbidity and disability. By mitigating the downstream consequences of mechanical brain injury on axons and neuronal connectivity, SARM1 inhibition could greatly improve functional outcomes. Small molecule SARM1 inhibitors seem well suited for targeting TBI due to the need for rapid attainment of therapeutic levels post-injury coupled with ease of administration relative to biologics. However, adequate blood-brain-barrier permeability would be imperative for central nervous system exposure. Structural modifications of lead peripheral neuropathy candidates or independent TBI programs could seek to enhance brain penetration. Still, local delivery methods directly to the injury site may prove necessary. Gene therapies seem less applicable to TBI given the spontaneous and acute nature of injuries. Though for moderate-severe injuries with high recurrence risk in certain professions (e.g. professional athletes), prophylactic gene therapy could prove beneficial. This would require one-time gene delivery to augment natural SARM1 expression with dysfunctional mutants that maintain immune cell viability but are poised to limit axon damage in future injury events. Safety considerations around neurological effects of long term SARM1 inhibition would need to be rigorously evaluated.
Unlike acute neurological injuries, neurodegenerative conditions like Alzheimer's and Parkinson's disease involve progressive neuronal dysfunction and loss over many years. However, Wallerian-like axon degeneration and death of axon projections contributes meaningfully to disease advancement. Though other parallel pathogenic processes also drive neurodegeneration, constraining this SARM1-mediated axonal destruction could help slow further advancement irrespective of additional disease drivers. Small molecule SARM1 inhibitors seem equally applicable for both acute and chronic neurological diseases due their ease of administration, ability to achieve sustained target inhibition, and distribution to affected neuronal tissues. However, blood-brain-barrier permeability would be essential for treating central nervous system disorders. Achieving exposure in relevant cell types may also prove challenging for certain neurodegenerative disorders that exhibit selective neuronal vulnerability. While gene therapy constitutes another attractive option for long term SARM1 inhibition in neurodegenerative diseases. One-time gene delivery of dysfunctional SARM1 variants or RNA interference could confer durable loss of enzymatic activity. Initial safety liabilities may be offset by slowing disease progression over time. Targeted gene delivery exclusively to affected neuronal subpopulations could also help avoid on-target effects in immune regulation. In assessing gene therapy for neurodegeneration, it warrants considering that while SARM1 promotes axonal degeneration in these disorders, its immune activity may help stimulate protective and regenerative pathways during disease progression via IL-6 and interferon signaling. Thus knockout approaches permanently ablating SARM1 function could prove detrimental long term compared to strand targeting or dominant negative methods permitting partial functionality.
Despite efficacy demonstrated already in preclinical models of neurological disorders, therapeutic SARM1 inhibition does carry risks around potentially detrimental on-target effects. While no overt phenotypes have been reported yet in constitutive SARM1 knockout animal models, its evolutionary conservation implicates possible physiological roles in metabolism, inflammation, or cell viability that could become relevant in humans with life-long inhibition. In the context of innate immunity, SARM1 functions as a negative regulator of TLR signaling and cytokine production by immune cells. Long term loss of these regulatory effects could hypothetically increase susceptibility to unchecked inflammation or infection. However, such phenotypes have not been evident thus far during periods of SARM1 ablation in animal models. In neuronal development, SARM1 contributes to processes like dendrite arborization, axon outgrowth and polarization. Whether long term therapeutic inhibition from childhood could subtly impair neurological development is controversial but warrants consideration to inform pediatric use. Monitoring for red flag impairment signs would help inform risk vs. benefit assessment.
For adult neurological disorders, the probable benefits seem to clearly outweigh hypothetical risks regarding ancillary SARM1 functionality. Halting axonal destruction and neuronal loss would provide pronounced gains in morbidity and mortality that outbalance theoretical infections or inflammation occurring at lower rates, especially given most patients are already in a disease compromised state. Risk calculus could differ for prophylactic inhibition in healthy individuals, but focusing initially on treating confirmed disease seems prudent while gathering additional safety data from long term animal models and initial clinical trials. Careful patient selection and monitoring would further help balance demonstrated efficacy gains against any theoretical safety issues emerging with prolonged SARM1 disruption.
SARM1 holds immense promise as a disease modifying therapeutic target to prevent irreparable axon destruction across an array of disorders spanning peripheral neuropathy, traumatic brain injury, and potentially neurodegenerative diseases. While small molecules, antibodies and gene therapy approaches each offer strengths and weaknesses, small molecules seem most poised as initial clinical candidates to treat peripheral neuropathies based on delivery, manufacturability and durability perspectives. Experimental therapeutics targeting SARM1 could profoundly change clinical outcomes for millions of neurological patients, and the progress already evident in preclinical animal models provides a high degree of confidence on probable efficacy with this approach. Further studies monitoring for theoretical long term safety issues will help refine the risk vs benefit profile, but the gains achievable by preventing permanent axon and neuron loss for devastating health conditions seems to clearly justify continued therapeutic pursuit of SARM1 inhibition.
