

Parkinson’s disease: proteinopathy or lipidopathy?
Parkinson’s disease: proteinopathy or lipidopathy?
Inventors & their inventions
Axial: https://linktr.ee/axialxyz
Axial partners with great founders and inventors. We invest in early-stage life sciences companies such as Appia Bio, Seranova Bio, Delix Therapeutics, Simcha Therapeutics, among others often when they are no more than an idea. We are fanatical about helping the rare inventor who is compelled to build their own enduring business. If you or someone you know has a great idea or company in life sciences, Axial would be excited to get to know you and possibly invest in your vision and company. We are excited to be in business with you — email us at info@axialvc.com
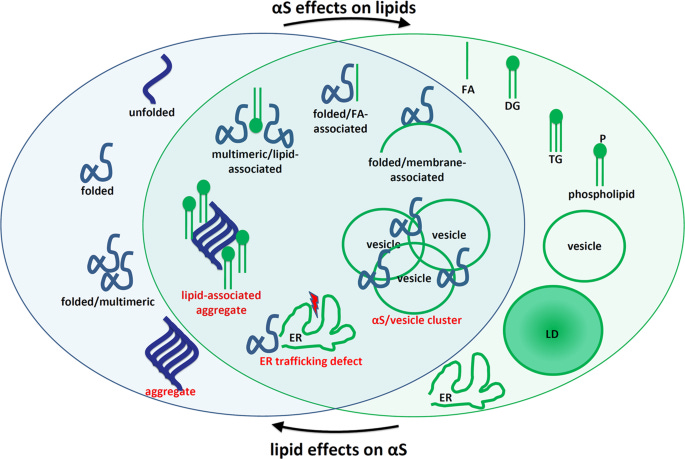
Parkinson's disease (PD) is a progressive neurodegenerative disorder characterized by motor symptoms, such as tremor, bradykinesia (slowness of movement), rigidity, and postural instability, as well as non-motor symptoms, including cognitive impairment, autonomic dysfunction, and sleep disorders. The primary pathological hallmark of PD is the accumulation of misfolded α-synuclein (αS) protein in intracellular inclusions called Lewy bodies and Lewy neurites, particularly in dopaminergic neurons of the substantia nigra pars compacta (SNpc) region of the midbrain.
The article presents a comprehensive review of the emerging evidence that suggests a pivotal role of lipid dyshomeostasis in the pathogenesis of PD and related synucleinopathies. The authors challenge the traditional view of PD as a proteinopathy, where the misfolding and aggregation of αS into fibrillar structures are considered the primary drivers of neurodegeneration. Instead, they propose that PD could be considered a "protein-induced lipidopathy," in which alterations in cellular lipid homeostasis are the primary neurotoxic events, and αS dyshomeostasis is the trigger.
1. αS is a lipid-binding protein: αS has been shown to interact with phospholipids and fatty acids through the formation of amphipathic helices, which facilitate its transient binding to cellular membranes. This interaction is proposed to be crucial for the physiological function of αS in regulating synaptic vesicle trafficking and neurotransmitter release.
2. Excess membrane binding of αS can lead to aggregation: While transient membrane binding is considered physiological, excessive or stabilized αS-membrane interactions can promote the formation of αS aggregates. Certain familial PD-linked mutations, such as E46K and A53T, have been shown to enhance αS-membrane binding, potentially contributing to aggregation and toxicity.
3. Membrane-associated αS aggregation in Lewy bodies: Recent findings from advanced imaging techniques have revealed that a significant portion of Lewy bodies, the pathological hallmark of PD, consists of membrane-rich structures coated with non-fibrillar αS, rather than solely fibrillar αS aggregates. This challenges the traditional view of Lewy bodies as amyloid-like fibrillar inclusions.
4. Bidirectional interplay between αS and lipids: The article proposes a bidirectional pathogenic loop between αS and lipids, particularly monounsaturated fatty acids (MUFAs) like oleic acid (OA). Excess αS, particularly membrane-associated monomers, can lead to increased levels of MUFAs, which in turn promote structural changes in αS, further exacerbating its aggregation and toxicity.
5. Genetics and patient data link PD to lipid pathways: Genome-wide association studies (GWAS) and postmortem analyses have identified several genes and proteins involved in lipid metabolism as risk factors for PD, including GBA (glucocerebrosidase), DGKQ (diacylglycerol kinase), and SREBF-1 (sterol regulatory element-binding protein-1). Additionally, alterations in specific lipid species, such as phosphatidylcholine (PC), phosphatidylethanolamine (PE), and phosphatidylinositol (PI), have been observed in PD patient brains and plasma.
6. Therapeutic potential of targeting lipid metabolism: Based on the proposed bidirectional αS-lipid interplay, inhibiting enzymes involved in MUFA biosynthesis, such as stearoyl-CoA desaturase (SCD), has emerged as a potential therapeutic strategy for PD. Preclinical studies have shown that SCD inhibition can mitigate αS aggregation and toxicity, potentially by counteracting the detrimental effects of elevated MUFAs on αS homeostasis.
The work presents a compelling argument for the involvement of lipid dyshomeostasis in the pathogenesis of PD and related synucleinopathies. By challenging the traditional view of PD as a purely proteinopathy and highlighting the intricate interplay between αS and lipid metabolism, the authors open up new avenues for understanding the disease mechanisms and developing novel therapeutic strategies.
However, it is important to note that the proposed "protein-induced lipidopathy" concept does not necessarily contradict the proteinopathy hypothesis but rather expands our understanding of the complex interplay between proteins and lipids in PD pathogenesis. It is possible that both proteinopathy and lipidopathy mechanisms contribute to the disease progression, and a vicious cycle of dyshomeostasis in protein folding and lipid metabolism may exist, as suggested by the authors.
Furthermore, while the article provides a comprehensive overview of the current knowledge, several areas require further investigation. For instance, the precise mechanisms by which αS interacts with specific lipid species and how these interactions contribute to aggregation and toxicity need to be elucidated in greater detail. Additionally, the role of other lipid-related pathways, such as those involving cholesterol and sphingolipids, in PD pathogenesis should be explored.
By highlighting the bidirectional interplay between αS and lipids, the authors offer new insights into the disease mechanisms and potential therapeutic targets. While further research is needed to fully understand the complexities of this interplay, the article paves the way for a paradigm shift in our understanding of PD and synucleinopathies, moving beyond the traditional view of these disorders as solely proteinopathies.
https://www.nature.com/articles/s41531-019-0103-7
