

Antibodies Aimed at Tau
Axial: https://linktr.ee/axialxyz
Axial partners with great founders and inventors. We invest in early-stage life sciences companies such as Appia Bio, Seranova Bio, Delix Therapeutics, Simcha Therapeutics, among others often when they are no more than an idea. We are fanatical about helping the rare inventor who is compelled to build their own enduring business. If you or someone you know has a great idea or company in life sciences, Axial would be excited to get to know you and possibly invest in your vision and company. We are excited to be in business with you — email us at info@axialvc.com
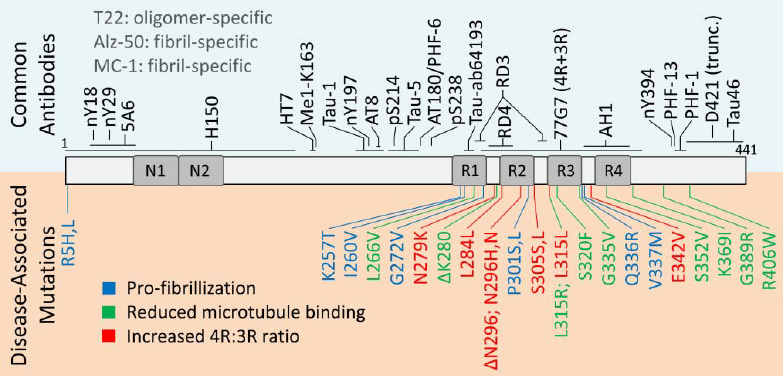
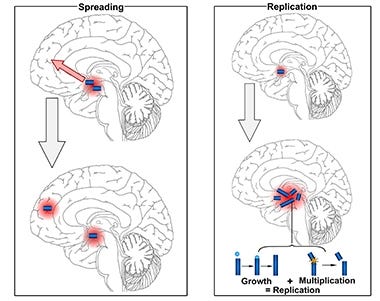
Tau is a microtubule-associated protein that plays a crucial role in the stabilization of neuronal microtubules and axonal transport. In several neurodegenerative diseases collectively known as tauopathies, including Alzheimer's disease (AD), tau becomes hyperphosphorylated, misfolds, and aggregates into insoluble fibrils that accumulate inside neurons. The spread of pathological tau between connected neurons is thought to drive neurodegeneration and cognitive decline in AD and other tauopathies.
Given the central role of pathological tau in disease pathogenesis, there has been great interest in developing therapies that can reduce levels of pathological tau species in the brain. One major therapeutic approach has been passive immunization with anti-tau monoclonal antibodies. The proposed mechanisms by which anti-tau antibodies could be therapeutically beneficial include:
1) Promoting clearance of extracellular pathological tau species from the brain and cerebrospinal fluid (CSF)
2) Blocking the spread of pathological tau between neurons
3) Disaggregating or preventing the aggregation of intracellular pathological tau species
Over a dozen anti-tau monoclonal antibodies have entered clinical trials for AD and other tauopathies over the past decade. However, the clinical results so far have been very disappointing, reminiscent of the repeated failures of anti-amyloid beta (Aβ) antibodies in AD trials prior to the recent approval of lecanemab and donanemab.
The antibodies in clinical trials have targeted different regions of the tau protein, including the N-terminal region, mid-domain, microtubule binding region, and C-terminal region. Several N-terminal targeting antibodies such as semorinemab, gosuranemab, tilavonemab, and zagotenemab have failed to show clinical efficacy in Phase 2 and 3 trials in AD and progressive supranuclear palsy (PSP), a primary tauopathy.
For example, the Phase 2 TAURIEL trial of semorinemab in 457 participants with early AD showed no slowing of cognitive decline despite evidence that the antibody increased plasma tau levels, suggesting peripheral sink effects. Gosuranemab, another N-terminal antibody, even worsened cognitive decline in the Phase 2 TANGO trial in 654 early AD participants, although it did reduce free N-terminal tau fragments in CSF.
One Phase 2 trial of semorinemab in 273 moderate AD patients (LAURIET) provided a hint of potential efficacy, with a 42% reduction in cognitive decline over 49 weeks compared to placebo. However, it failed to improve functional abilities or other secondary endpoints.
Similarly, the Phase 2 trials of tilavonemab and gosuranemab in PSP failed to show any benefits on disease progression as measured by the Progressive Supranuclear Palsy Rating Scale, despite evidence of target engagement based on reductions in free N-terminal tau in CSF.
The consistent failures of N-terminal tau antibodies prompted a shift in focus to antibodies targeting other regions. Antibodies binding the mid-domain or microtubule binding regions are hypothesized to better prevent the spreading of pathological tau species between neurons. Several such antibodies (e.g. sembranemab, bepranemab) have now advanced into early stage clinical testing.
One key question is whether simply reducing levels of extracellular pathological tau is sufficient for clinical benefit, or if antibodies need to clear intraneuronal tau aggregates as well. Preclinical studies suggest the latter may be necessary. For example, active immunization with tau antibodies that can cross the blood-brain barrier and enter neurons provided greater therapeutic benefits in tau transgenic mice compared to peripheral immunization.
Another key issue is whether the toxic species are soluble tau oligomers/pre-fibrils or insoluble fibrillar tau aggregates. Most antibodies have focused on binding soluble extracellular species, but some newer candidates aim to selectively target insoluble tau fibrils/aggregates which may be the primary drivers of neurodegeneration.
Additionally, the precise conformation of pathological tau targeted is likely important, as tau can misfold into diverse structures (e.g. different strains) that may vary across tauopathies and even within brain regions of the same disease. Some of the newer generation antibodies claim greater selectivity for disease-specific pathological tau conformations over normal physiological tau.
Optimal timing of intervention is also critical, as removing pathological tau late in the disease course when substantial neuronal loss has already occurred may have limited clinical benefits. Most trials have focused on early symptomatic AD stages, but future prevention trials in preclinical AD cases based on tau PET imaging or other biomarkers may be more effective.
Another strategy could be combining anti-tau therapies with anti-amyloid therapies, as preclinical data indicates synergistic benefits. However, failed trials of dual Aβ/tau vaccines suggest this combination approach may face hurdles as well.
Overall, while the initial anti-tau antibody trials have been largely disappointing, the field remains optimistic that therapies selectively targeting the most toxic tau species and strains with improved brain exposure and at the optimal therapeutic window can still prove effective for treating AD and other tauopathies. Intense efforts are underway to develop such next-generation, selective anti-tau immunotherapies to test this hypothesis.
Some key lessons from the initial clinical failures include: 1) Reducing extracellular pathological tau alone may be insufficient; therapies that can clear intraneuronal aggregates may be required. 2) N-terminal tau antibodies may interfere with normal physiological tau functions. 3) The toxic tau species/strains being targeted need to be carefully selected and validated. 4) Optimal therapeutic windows based on accurate tau biomarkers are crucial to intervene before widespread neurodegeneration occurs.
The repeated failures highlight that the underlying mechanisms linking pathological tau to neurodegeneration and cognitive deficits are still not fully understood. Some have questioned whether extracellular tau is merely a bystander phenomenon, while intracellular loss of normal tau function at microtubules is the primary driver of toxicity.
There are also concerns that reducing total tau levels below a critical threshold, including physiological tau species, could actually worsen neurodegeneration. As with anti-amyloid therapies,excessive depletion of physiological tau should be avoided. Future trials need to carefully monitor for this potential adverse effect.
While the initial anti-tau antibody trials have been largely disappointing so far, mirroring the troubled history of anti-amyloid immunotherapies, the field remains highly active in developing improved next-generation candidates. Continued advances in biomarker development, improved antibody selectivity and brain exposure, optimal therapeutic timing, and combination therapies may eventually unlock the potential of this approach. However, the failures also raise the possibility that fundamental assumptions about the role of pathological tau in driving neurodegeneration may need to be re-evaluated.
